Rett syndrome is a rare and devastating neurological disorder caused by a mutation on the X chromosome, primarily affecting girls. These affected girls exhibit impaired cognitive abilities and autistic behaviors, often from their first year of life, due to a mutation in the MECP2 gene on one of their X chromosomes. In contrast, boys, who have only one X chromosome, almost never carry this mutation. Is this an extreme case of a “sexist mutation,” or have males found a way to protect their unique X chromosome?
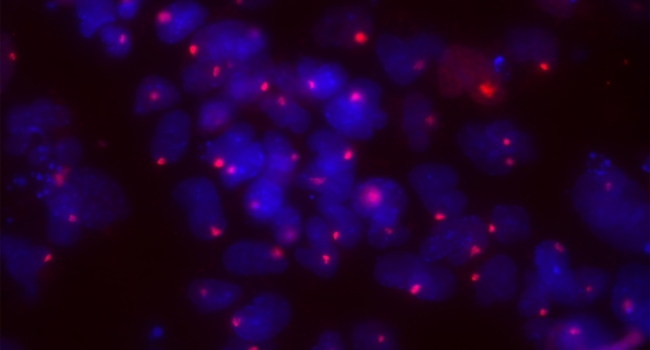
The reality is neither. The mutation in the MECP2 gene likely arises as frequently in male fetuses as in female fetuses. However, female fetuses have a genetic advantage: with two X chromosomes, only about 50% of their cells express the mutated gene, while the other 50% express the healthy gene. This situation results from a balancing process called X chromosome inactivation, where one of the two X chromosomes in females is randomly silenced to achieve equilibrium in X-linked gene expression between sexes. While the fetus may not develop ideally, it can still survive, and the girl is born with a severe disability.
On the other hand, male fetuses, having only one X chromosome, express it in all their cells. Without a compensating healthy X chromosome, the fetus cannot develop properly and rarely survives birth. Thus, very few boys are born with Rett syndrome because affected fetuses typically do not make it that far. Counterintuitively, females have a mechanism to protect themselves from the MECP2 mutation on the X chromosome.
Understanding the mechanisms underlying X chromosome inactivation and reactivation could lead to life-saving treatments. Since females silence about 50% of the functional copy of any X-linked genes during this process, they are a living mosaic of cells expressing either the healthy (wild-type, WT) or mutated copy of a protein. Cells expressing the mutated form may not function as well as those expressing the WT copy. Even in cells with the mutated gene, there exists a “dormant” healthy copy that can potentially be reactivated to alleviate the disease. The condition often results from a reduced dose of functional protein, which then cannot fulfill its biological role.
The X chromosome is essential for brain development, with many X-linked genes regulating brain development and function. Therefore, reactivating the inactive X chromosome could be crucial for treating several genetic diseases, from poorly characterized disorders like CDKL5 and Fragile X syndromes to more well-known conditions like Rett syndrome. These diseases are associated with autistic behaviors, severe intellectual disabilities, and neuropsychiatric traits.